




The main goal of the PerMedCoE project is to provide an efficient and sustainable roadmap to scale up a collection of relevant computational biology tools (known as core applications), focusing on solutions that could leverage the next generation of HPC exascale platforms. These core applications implement different modelling approaches that allow researchers to perform multi-scale simulations of complex biological systems such as healthy tissues and tumours. To achieve its goals, PerMedCoE will contribute to an extensive refactoring of the core applications, thereby producing a considerable increase in speed and scalability. As a consequence, the new developments will support more complex and realistic cellular scenarios that usually demand a higher number of cells in multi-scale modelling simulations, more complex signalling pathways to properly address cell-type specificity, or the implementation of larger-scale models in metabolic studies, among others. At the end of the project, we will provide a rich computational framework to translate omics analyses into actionable models of cellular functions, with great importance to build patient-specific models in the context of emerging paradigms such as targeted therapy or personalised medicine.
The selected core applications represent a set of open-source reference solutions to model and simulate the activity of signalling pathways and metabolic networks using complementary approaches. In this context, one of the most important challenges of our project is that we are dealing with already existing tools, developed in a set of very different programming languages, such as C++ or R. This circumstance expands the repertoire of strategies that we need to follow to efficiently port the source code to HPC environments, prioritising, in this case, the use of specific protocols for parallel and distributed computing, such as OpenMP or MPI, or opting to use programming languages such as Julia, specially designed to work on this type of contexts.
Our current work on PhysiCell is an illustrative example of the PerMedCoE approach. PhysiCell is a physics-based tool designed to simulate the evolution and dynamics of a heterogeneous population of cells, such as tumour masses. To this end, PhysiCell implements an agent-based paradigm that efficiently models cell-specific behaviours, including different features such as cell growth, proliferation or motility. These implemented features provide a powerful framework capable of simulating a wide variety of real cellular scenarios, often considered as a virtual laboratory.
PhysiCell is fully written in C++ language and relies on the BioFVM partial differential equations solver, which allows the simulation of different substrates throughout the entire extracellular space. The PhysiCell roadmap focuses on refactoring the official version to use the MPI protocol. MPI is a gold standard technology that allows the computational load of a given software to be efficiently distributed among different computers connected to the same network. Also, memory resources are managed in a similar distributed manner, of particular interest in problems where memory requirements exceed the available capacity of individual machines. In our case, the main strategy for scaling up PhysiCell is centred on dividing the physical space where the cells are located into different subdomains. Then, each subdomain is specifically managed by one individual MPI node, allowing to scale up individual simulations at a much higher scale.
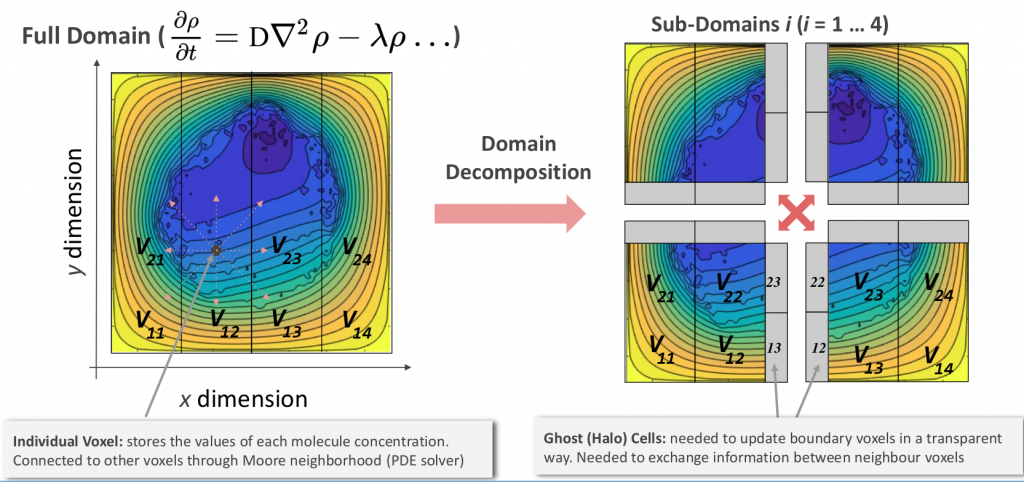
Representative diagram of the MPI-based solution implemented in PhysiCell © Miguel Ponce De León (BSC)
In a similar way, the MaBoSS tool will be re-engineered with an MPI-based solution. MaBoSS is a simulation tool for continuous/discrete-time Markov processes applied to a Boolean network. It parameterises the models by introducing transition rates between proteins, thus extending the functionality of purely Boolean models. In practice, the tool stochastically simulates a large number of network trajectories intending to find and characterise the different stable states that the network could reach. The current MaBoSS version, written in C++, parallelise this work among different CPUs by using POSIX threads, which will be complemented by the use of the MPI protocol, thus allowing the different simulated trajectories to be also distributed among different machines.
A different strategy has been followed for scaling up COBRA toolbox. This tool implements a set of standard methods in systems biology to bring biochemical reaction-level computational models of living organisms. The available methods, mainly based on solving constrained linear problems, will be adapted to decompose the work in a set of different subtasks that will be parallelized across different machines. To this end, we are developing a new software package based on the Julia programming language, which includes a suitable internal data serialization format for storing, manipulating, and exchanging the model information within the nodes. Therefore, our approach will provide a complete framework to run very large models of cells and organisms on pre-exascale HPC environments.
Also, CellNOpt will require a specific solution. CellNOpt is an ecosystem of tools for the modelling of cell signalling. These tools exploit different optimisation techniques for inferring signalling networks, combining current biological knowledge to provide cell type- or tissue-specific models. In order to increase the scalability of these tools, we will conduct an extensive benchmark to compare different general-purpose optimisers, including the most popular Linear Programming solvers such as C-PLEX or Gurobi. Consequently, we will evaluate which solutions are more suitable to a parallel and distributed computing environment such as HPC pre-exascale technologies, with great impact in other areas of our projects, since these generic pieces of code could be easily integrated to solve different kinds of scientific problems.
Finally, one of the most interesting objectives of our project is the integration of different core applications into a single multi-scale modelling solution. This strategy will produce more realistic simulations, since known molecular pathways describe the inherent response of cells to different extracellular stimuli and, therefore, the main features that regulate their life cycle. A good example of this strategy is represented by the PhysiBoSS tool, which describes the integration of PhysiCell + MaBoSS. This tool defines cell behaviour based on existing signalling pathways, modelled by the stochastic approach of MaBoSS. Following this idea, we will suggest future developments including metabolic models, and their combination with signalling networks. We envision that such approaches will be the future of personalised medicine.
Author: José Carbonell (BSC)